Simulation
Unity simulation
To confirm our design concept, we developed a simulation software using the physics engine Unity [1], which allows us to explore the space of design parameters and predict the behavior of molecules in nano-scale.
1.Model
Since it takes too much computational cost to represent the 3D model of the monomer in detail, we have to made necessary simplifications in the simulation.
- Simulation space is set as a cube with periodic boundaries to reduce the computing time. Hundreds of monomers placed randomly in a space with determined concentration
- To simulate the collision efficiently, we modify the position of monomer, which goes out of the boundaries to satisfy the periodical boundary. When two monomers collide, we erased one of the object and generate a new object with correct length instead of combining two objects. It is possible to separate the object into two pieces.
- The monomers are simplified as cylinders, which is cost-efficient for the physics engine.
- The stacking of the monomers takes place as the collisions among the cylinders.
2.Interface
The cylinders collide mutually based on Brownian motion. We created a user interface with several scripts:
- To adjust the space size to fit with the desired concentration.
- To modify the velocity and angular velocity of each monomer.
- To output the result into an appropriate file format.
3.Results
In our simulation, the Brownian motion of each monomer and the stacking behavior among them were reproduced. The results correspond to the temperature at 8 °C and 250 nM concentration of DNA origami in a span of 8 seconds. The graph (Fig. 3) and the histogram (Fig. 4) show that monomer concentration decreased while the multimers such as dimers and trimers increased overtime.
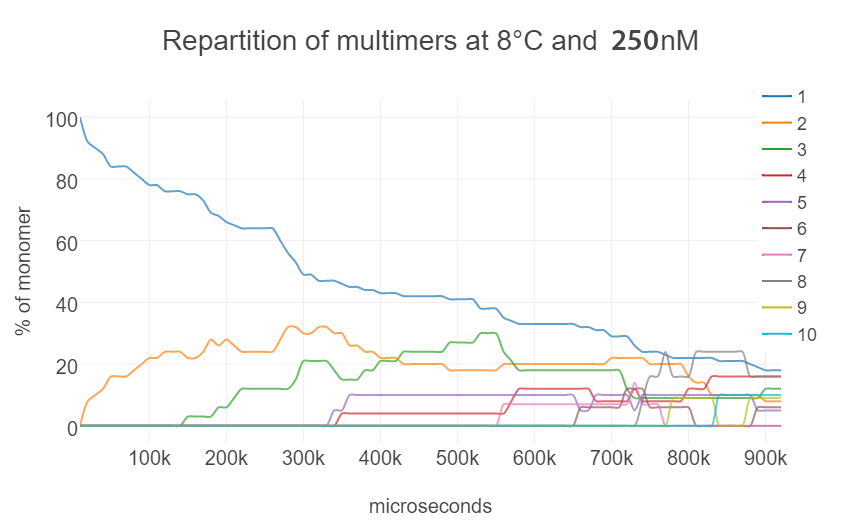
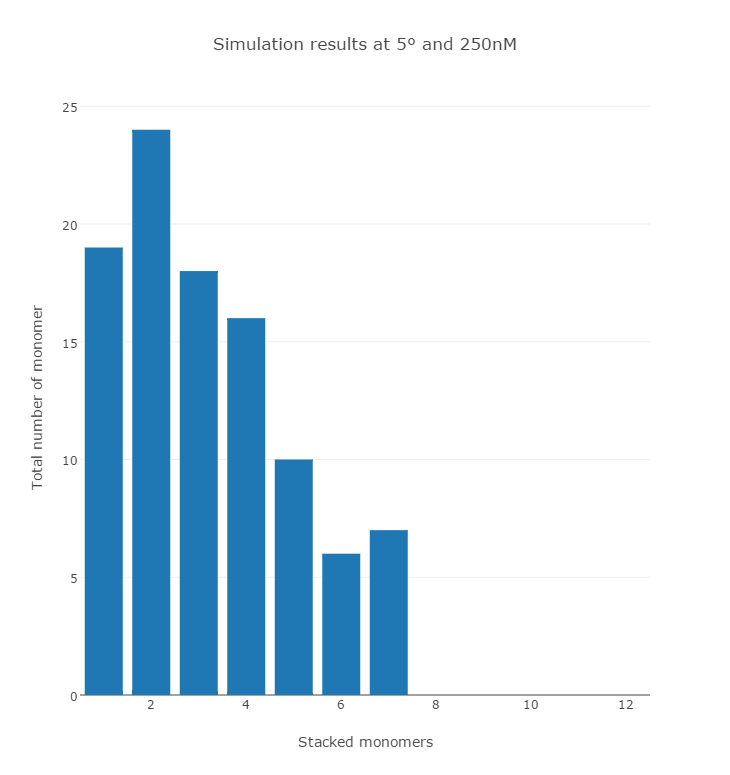
There are still some unrealistic motions due to the limitation of the Unity engine, since this engine is not made to work on the molecular scale, it cannot handle colliding objects at high speed. We plan to improve the simulation in the following ways: (1) Introducing CUDA to reduce the computational time. (2) Employing parallel processing to compute the time development in parallel to obtain average data.
If you are interested in this program, please visit here.
oxDNA simulation
To investigate the thermal fluctuation of the monomer in solution, we carried out a coarse-grained molecular dynamic simulation. The result of oxDNA [2] (Moive 2, Moivie 3) shows that the shaft and the cylinder rotate independently which means the origami structure formed correctly as designed.
Reference
- Unity: http://unity3d.com/
- oxDNA: https://dna.physics.ox.ac.uk
Jonathan P. K. Doye, Thomas E. Ouldridge, Ard A. Louis, Flavio Romano, Petr Sulc, Christian Matek, Benedict E. K. Snodin, Lorenzo Rovigatti, John S. Schreck, Ryan M. Harrison, William P. J. Smith. Coarse-graining DNA for simulations of DNA nanotechnology. Physical Chemistry Chemical Physics, 15, 20395-20414 , 2013